Author : Eeva-LiisaEskelinen PaulSaftig
Abstract
Autophagy delivers cytoplasmic material and organelles to lysosomes for degradation. The formation of autophagosomes is controlled by a specific set of autophagy genes called atg genes. The magnitude of autophagosome formation is tightly regulated by intracellular and extracellular amino acid concentrations and ATP levels via signaling pathways that include the nutrient sensing kinase TOR. Autophagy functions as a stress response that is upregulated by starvation, oxidative stress, or other harmful conditions. Remarkably, autophagy has been shown to possess important housekeeping and quality control functions that contribute to health and longevity. Autophagy plays a role in innate and adaptive immunity, programmed cell death, as well as prevention of cancer, neurodegeneration and aging. In addition, impaired autophagic degradation contributes to the pathogenesis of several human diseases including lysosomal storage disorders and muscle diseases.
5. Physiological functions of autophagy
5.1. Stress response and housekeeping function
The role of autophagy as a survival mechanism during short-term amino acid starvation is well documented. Macroautophagy is induced by starvation of serum and amino acids; autophagosomes can be detected already after 15–30 min of starvation [55]. During long-term starvation, chaperone-mediated autophagy increases and macroautophagy decreases [80], [81]. Yeast strains defective in autophagy do not survive nitrogen starvation [38]. Knockout mice deficient in one of the central autophagy proteins, Atg5, show that autophagy is indispensable for the energy metabolism immediately after birth [82]. Atg5 knockout mice die of starvation one day after birth.
In muscle and heart cells, autophagy seems to have a special housekeeping role in the turnover of cytoplasmic constituents including mitochondria. This is revealed by myopathy and cardiomyopathy in patients and mice possessing a defective autophagic degradation due to deficiency of the lysosomal membrane protein LAMP-2 [83], [84], [85]. LAMP-2 deficiency is described in detail later in this review. The importance of autophagy for the heart muscle is supported by a study showing that heart-specific loss of the autophagy protein Atg5 causes cardiomyopathy in mice [86]. Evidence has been published suggesting that damaged mitochondria might be autophagocytosed selectively in a process termed mitophagy [87]. Mitochondria are the major source of reactive oxygen species in cells. Interestingly, reactive oxygen species are necessary for the signal transduction pathway initiating starvation-induced autophagy [88].
It was proposed long ago that autophagy has a role in growth regulation, as suggested by decreased autophagy during growth of the kidney after unilateral nephrectomy [89]. Inducible knockdown of the autophagy protein Atg5 in cell culture shows that autophagy negatively controls cell size [90]. Similar result was observed in Drosophila fat body over-expressing the autophagy protein Atg1 [91].
Autophagy contributes to intracellular quality control and housekeeping, especially in turnover of aggregate-prone proteins. Prevention of autophagy by conditional knockout of atg7 leads to accumulation of ubiquitinated protein aggregates in mouse tissues [92]. Tissue-specific knockout of autophagy proteins in the central nervous system causes accumulation of ubiquitin-positive protein aggregates and neurodegeneration in mice [93], [94]. Further, enhanced autophagy reduces the toxicity of the Huntingtin protein aggregates that accumulate in Huntington disease [95]. Autophagy may prevent aggregate formation by degrading the proteins as monomers, oligomers, or after aggregate formation [96]. It is not clear at present whether aggregated proteins are segregated preferentially, or whether they are removed via unspecific autophagic uptake of cytoplasm. Two proteins have been proposed to function during the uptake of protein aggregates: Alfy and p62 [97], [98]. p62 binds to both ubiquitin-conjugated aggregate-prone proteins and the autophagosome protein LC3 [99], which suggests that it could selectively recruit autophagy machinery to the aggregates and enhance their autophagic clearance.
In addition to removal of cytoplasmic aggregate-prone proteins, autophagy also contributes to the quality control in the ER. Unfolded protein response induces autophagy, and this induction is beneficial for cell survival [100], [101], [102].
Autophagic degradation is also needed for early embryonic development. A recent study shows that autophagy-defective mouse eggs fertilized with autophagy-defective sperm, failed to develop beyond the four and eight cell stages [103]. The authors suggest that autophagy may be needed in the preimplantation embryos for protein recycling, production of amino acids for protein synthesis or substrates for energy production, or for removal of obsolete maternal factors.
5.2. Innate and adaptive immunity
Autophagy contributes to both innate and adaptive immunity [104], [105]. In some cases autophagy can protect cells against intracellular pathogens. Induction of autophagy during Herpes simplex virus infection, and localization of viral particles inside autophagic vacuoles, were proposed to indicate that autophagy acts as a host-defense mechanism in infected cells [59]. The Herpes virus virulence protein, ICP34.5, inhibits autophagy, suggesting that the virus has developed a way to prevent the autophagic defense of the host cell. Autophagy may also help cells to defend against intracellular bacteria [106]. Sequestration of intracellular Group A Streptococci in autophagosome-like structures protects cells against the bacteria [107]. Mycobacterium tuberculosis is normally able to survive inside macrophages by preventing the fusion of phagosomes with lysosomes. Surprisingly, induction of autophagy bypasses the maturation defect, leading to phagolysosome formation and bacterial killing [108].
Macroautophagy also contributes to antigen presentation. Major histocompatibility complex (MHC) class II molecules present products of lysosomal proteolysis to CD4(+) T cells. Extracellular antigen uptake is considered to be the main source of MHC class II ligands. However, it was demonstrated that in MHC class II-positive cells, including dendritic cells, B cells, and epithelial cells, autophagosomes continuously fuse with multivesicular MHC class II-loading compartments [109]. This pathway is of functional relevance, because targeting of the influenza matrix protein 1 to autophagosomes enhances its MHC class II presentation to CD4(+) T cells. Thus it seems that macroautophagy efficiently delivers cytosolic proteins for MHC class II presentation and can improve helper T cell stimulation.
5.3. Cell death
Autophagy also seems to have roles in programmed cell death [110], [111]. Type II programmed cell death, or autophagic cell death, was originally described in mammary carcinoma cells [112], [113]. Autophagy proteins were shown to be necessary for cell death under certain conditions, such as in apoptosis-defective cells [114], [115], [116]. In this scenario autophagy is needed for the execution of cell death. Under other conditions, such as nutrient starvation, autophagy protects cells against apoptosis by providing nutrients [117], [118], [119]. The regulation of apoptosis and autophagy are linked via the antiapoptotic protein Bcl-2. Bcl-2 inhibits Beclin 1-dependent autophagy by binding to Beclin 1 and preventing its association with Vps34 [52]. This anti-autophagy function of Bcl-2 was proposed to help maintain autophagy at levels that are compatible with cell survival, rather than cell death.
Lipids may also regulate autophagy and its outcome to the host cell. Ceramide and sphingosine 1-phosphate, a metabolite of ceramide, both induce autophagy in mammalian cells [120]. The outcome on cell survival is however different: ceramide promotes cell death, while sphingosine 1-phosphate increases cell survival. Ceramide is part of the signaling cascade initiated by chemotherapy, while sphingosine 1-phosphate is part of the signaling cascade initiated by starvation. Beclin 1 level and the autophagy response are stronger during ceramide signaling [121].
Autophagy has functions in cell death during development [122]. atg genes are necessary for the clearance of apoptotic cells during embryonic development in mice [123]. Autophagy contributes to dead-cell clearance during programmed cell death by maintaining cellular energy levels in the dying cells, thereby allowing the generation of cell surface and secreted signals that then promote engulfment of cell corpses by neighboring cells. Autophagy is also indispensable for the execution of certain types of cell death during development. The degradation of Drosophila salivary glands by type II programmed cell death depends on autophagy [73].
5.4. Aging and longevity
Finally, autophagy also contributes to longevity [124]. Reduced caloric intake increases longevity in several animal species. Increased autophagic turnover of cytoplasmic constituents including mitochondria was shown to contribute to the longer life in the dieting animals [125]. Further evidence that autophagy contributes to longevity come from Caenorhabditis elegans mutants possessing a defective insulin receptor (daf2 mutant), which live longer than control worms. The increased lifetime of these mutant worms depends on a functional autophagic pathway [126]. Moreover, knockdown of autophagy gene products including Atg7 and Atg12 were shown to shorten the lifespan of both wild type and daf2 mutant C. elegans [127]. Further, promoting basal levels of autophagy in the nervous system of adult Drosophila enhances longevity of the flies [128]. Together these studies give strong support for a role of autophagy in the prevention of aging.
Fig. 6 summarizes the physiological functions of autophagy described above.

6. Autophagy and disease
6.1. Cancer
Impaired autophagy contributes to cancer development [2], [129], [130]. Beclin 1 is monoallelically deleted in a large proportion of human breast and ovarian cancers. Over-expression of Beclin 1 in a breast cancer cell line increases autophagy and decreases the growth and tumorigenicity of these cells [131]. Mice with heterozygous deletion of Beclin 1 have less autophagy and more tumors than control mice [132], [133]. Further, the other autophagy-promoting components of the Beclin 1/Vps34 complex, UVRAG and Ambra 1 (Fig. 4), are also tumor suppressors [48], [49]. Moreover, knockout of Bif-1, also part of the Beclin 1 complex, significantly enhances the development of spontaneous tumors in mice [50]. On the other hand, binding of the proto-oncogenic proteins Bcl-2 or Bcl-XL to Beclin 1 inhibit autophagy [51], [52]. In addition to the Beclin 1 complex, other tumor suppressors also enhance autophagy. PTEN is a phosphatase that decreases the concentration of class I PI3 kinase product and enhances autophagy [72]. PTEN is also a tumor suppressor [134]. Further, the activities of Ras and class I PI3-kinases inhibit autophagy and promote cell growth. Ras is mutated and class I PI3 kinases are upregulated in many cancers [135], [136].
The results described above show that autophagy contributes to the prevention of tumorigenesis. Impaired autophagy can contribute to tumor formation via impaired regulation of cell growth, and/or via decreased cell death. In addition, it was shown that failure to sustain metabolism via autophagy results in increased DNA damage. This chromosomal instability increases tumor progression [137].
In advanced cancers, autophagy may have the opposite effect on the tumor development. Autophagy can benefit the progression of the tumor because it can provide nutrients during starvation [129], [130], [138]. In addition, autophagy was recently shown to improve the survival of p53-deficient cancer cells under starvation or hypoxic conditions [139]. These findings suggest that autophagy inhibition, rather than stimulation, might be beneficial in treatment of advanced cancer.
6.2. Neurodegeneration
Many age-related neurodegenerative diseases are characterized by the accumulation of ubiquitin-positive protein aggregates in affected brain regions. These misfolded, aberrant proteins can disrupt neuronal function and cause neurodegeneration. As described earlier, autophagy is necessary for the clearance of aggregate-prone proteins that are toxic especially for post-mitotic cells like neurons [130]. Tissue-specific knockout of the autophagy genes in neurons causes a massive accumulation of ubiquitin-positive protein aggregates and neurodegeneration in mice [93], [94], indicating that autophagy is needed for the constitutive clearance of aggregate-prone proteins. Autophagy was recently shown to enhance the clearance of Huntingtin, mutant tau, synphilin 1 and α-synuclein, but not AIMP2 (p38) and mutant desmin [140]. This study indicates that autophagy is not able to degrade all protein aggregates. However, the role of autophagy has been demonstrated in Huntington’s disease, caused by mutations in Huntingtin, and familial Parkinson’s disease, caused by mutations in α-synuclein. Enhanced autophagy in animal models of these diseases improves clearance of the aggregated proteins and reduces the symptoms of neurodegeneration [95], [141].
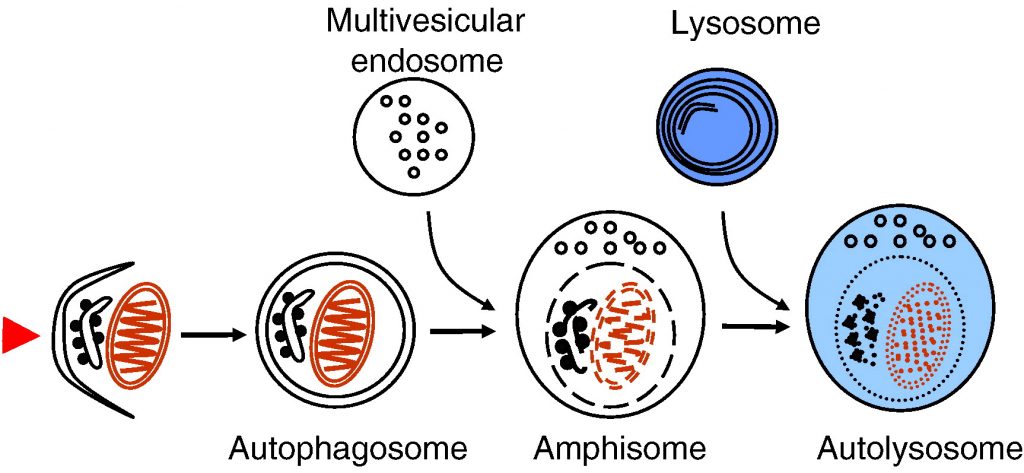
ESCRT complexes are necessary for the biogenesis of multivesicular endosomes. As described earlier, multivesicular endosomes are necessary for the maturation of autophagosomes into degradative autolysosomes. Mutations in ESCRT III subunits CHMP2B or mSnf7-2 are associated with two neurodegenerative diseases, frontotemporal dementia and amyotropic lateral sclerosis. Both diseases are characterized by abnormal ubiquitin-positive protein deposits in affected neurons. Cell lines and fruit flies depleted of CHMP2B or mSnf7-2 show decreased autophagic degradation, increased levels of ubiquitin-positive aggregates and increased neurodegeneration [27], [29].
Alzheimer’s disease is characterized by the accumulation of extracellular amyloid plaques in the brain. These plaques consist of aggregated β-amyloid (Aβ) peptide. Autophagy was proposed to contribute to the production of Aβ. Autophagic compartments containing both amyloid precursor protein and Aβ accumulate in dystrophic neurons in Alzheimer brain [142], [143]. Purified autophagic vacuoles contain all necessary constituents for Aβ production [142], and autophagic compartments were identified as a major reservoir of intracellular Aβ in the brain of Alzheimer patients and mouse models. The primary cause for the increased accumulation of autophagic compartments in Alzheimer’s disease was recently suggested to be their retarded maturation to autolysosomes [144].
A recent study, however, challenges the idea that autophagy contributes to the pathogenesis of Alzheimer’s disease. Beclin 1 was shown to be decreased in affected brain regions of patients with Alzheimer disease early in the disease process [145]. Heterozygous deletion of Beclin 1 in mice decreased neuronal autophagy and resulted in neurodegeneration. Transgenic mice expressing human amyloid precursor protein have been used as a mouse model for this disease. Genetic reduction of Beclin 1 expression increased intraneuronal Aβ accumulation, extracellular Aβ deposition, and neurodegeneration [145]. Increasing Beclin 1 levels by lentiviral expression reduced both intracellular and extracellular amyloid pathology in these transgenic mice. This study suggests that decreased, not increased, autophagy promotes Alzheimer’s disease progression. Further, enhancing autophagy by increasing Beclin 1 levels may have therapeutic potential in this disease.
6.3. Autophagy and lysosomal storage diseases
Niemann–Pick type C is a neurodegenerative lipid storage disorder characterized by a disruption of sphingolipid and cholesterol trafficking caused by mutations in either of two genes, npc1 and npc2. The disease produces cognitive impairment, ataxia and death, often in childhood. Cells deficient in npc genes show increased expression of Beclin 1 and LC3-II, the autophagosome-specific form of LC3, suggesting autophagy is induced [146]. Increased levels of LC3-II have also been observed in npc-deficient brain tissue [147]. npc-Deficient cerebellar Purkinje neurons undergo a cell death that was proposed to depend on autophagy [148], suggesting increased autophagy may be harmful for neurons in NPC patients.
Most lysosomal storage diseases are caused by deficiencies of lysosomal hydrolases, leading to accumulation of undegraded substrate and other material in the lysosomal compartment. Lysosomal accumulation of substrates can also affect autophagosome–lysosome fusion. Autophagosomes accumulate in brain and isolated cell lines of mouse models of two lysosomal storage diseases associated with severe neurodegeneration, multiple sulfatase deficiency and mucopolysaccharidosis type IIIA [149]. Significantly reduced colocalization of the lysosomal membrane protein LAMP-1 with the autophagosome marker LC3 indicates that the fusion of lysosomal compartments with autophagosomes is impaired. In addition, cell lines isolated from these mice have decreased ability to degrade aggregate-prone proteins and show accumulation of polyubiquitinated proteins and non-functional mitochondria. Thus, neurodegeneration observed in many lysosomal storage diseases may be at least partially due to impaired autophagic degradation, which is particularly vital for neurons.
6.4. Autophagy and muscle disorders
Autophagic vacuoles are a frequent feature in numerous muscular disorders. Such a pathological situation can be observed in patients suffering from Danon disease, an inherited disease resulting from null mutations in the lysosomal membrane protein LAMP-2 [83]. LAMP-2 deficiency leads to a fatal cardiomyopathy and myopathy sometimes associated with mental retardation [150]. Accumulation of autophagic vacuoles in the heart and skeletal muscle are hallmarks of the disease [84]. Studies in LAMP-2 deficient mice revealed in part similar findings [85]. Fifty percent of these mice die at an early postnatal age with massive accumulation of autophagic vacuoles in several tissues including liver, pancreas, spleen, kidney, lymph nodes, neutrophilic leukocytes, skeletal muscle, and heart. Autophagic vacuoles containing single mitochondria were frequently observed in cardiomyocytes [151] indicating that mitochondria are a main target for autophagic degradation in muscle tissues. These cellular alterations lead to a reduced contractility and an increased size of the heart in LAMP-2 knockout mice. This is in agreement with the finding that cardiomyopathy is the hallmark in Danon disease patients [83]. Biochemical and electron microscopy studies reveal that a retarded consumption, rather than increased formation, of autophagic vacuoles leads to their accumulation [152]. LAMP-double deficient fibroblasts lack both LAMP-2 and the structurally related LAMP-1 protein. These cells show a defect in the final maturation steps of late autophagic vacuoles, involving retarded fusion with lysosomes [33], [119]. Interestingly, recruitment of the small GTPase Rab7 to autophagosomes is retarded in these cells [32].
Inhibition of lysosomal fusion using hydroxy-chloroquine causes similar vacuolar alterations and myopathies to Danon disease, confirming the important role of lysosome–autophagosome fusion for muscle cell physiology [153]. Impaired autophagosome maturation may also be related to other types of diseases such as X-linked myopathy with excessive autophagy, infantile autophagic vacuolar myopathy, adult-onset autophagic vacuolar myopathy with multiorgan involvement, and X-linked congenital autophagic vacuolar myopathy [154]. The molecular defects in these disorders are still unknown.
Although altered autophagy has been observed in various heart diseases, including cardiac hypertrophy and heart failure, it remains unclear whether autophagy plays a beneficial or detrimental role in these diseases. As mentioned earlier, tissue-specific deletion of atg5 in heart causes cardiac hypertrophy and contractile dysfunction [86]. In addition, increased levels of ubiquitinated proteins and abnormal mitochondria are found, especially after treatment with pressure overload or β-adrenergic stress. This suggests that autophagy is needed in the heart to ensure the availability of sufficient energy substrates and to control cardiomyocyte size and global cardiac structure and function.
6.5. Common aspects in autophagy and phagocytosis
As described above, autophagy plays a role in innate immunity against intracellular pathogens [104], [105] by clearing microbes directly via ingestion into autophagosomes for subsequent degradation in autolysosomes [108], [155]. Similar to the process of intracellular defense, phagocytosis is an evolutionary conserved mechanism involved in the removal of extracellular organisms. Interestingly, it was found that the phagocytic and autophagic pathways are linked. Toll like receptor (TLR) activation triggers the recruitment of autophagy proteins LC3, Atg5 and Atg7 to the phagosomal pathway. Before these events Beclin 1 and class III PI3-kinase activity are found in phagosomes [156]. These autophagy-specific proteins are recruited to the phagosome, while almost no classical autophagosomes are observed in the cells. Phagosome fusion with lysosomes is then initiated, leading to acidification and killing of the ingested organisms. Thus, engaging the autophagy pathway via TLR signaling (especially TLR7 through its binding to single-stranded RNA) enhances phagosome maturation and destruction of pathogens [157]. This underscores the intimate link between autophagy and phagocytosis.
As mentioned earlier, M. tuberculosis is able to survive inside macrophages by preventing the fusion of phagosomes with lysosomes, but induction of autophagy bypasses the maturation defect, leading to phagolysosome formation and bacterial killing [108]. Autophagy induction induces the localization of Beclin 1 and LC3 to phagosomes, suggesting the phagosomes are diverted to an autophagosome-like compartment that is then able to fuse with lysosomes.
The association between autophagy and phagocytosis is also underlined by studies with cells lacking either one or both LAMPs [33]. As described above, autophagosome–lysosome fusion is impaired in LAMP-double deficient cells [32]. Whereas macrophages and fibroblasts from LAMP-1 or LAMP-2 single-deficient mice display normal fusion of lysosomes with phagosomes, in LAMP-double knockout fibroblasts phagosomes are unable to recruit late endosomal/lysosomal markers and phagocytosis is arrested prior to the acquisition of Rab7 [158]. Interestingly, the maturation of Neisseria-containing phagosomes is also disturbed and cells lacking both LAMP proteins fail to kill the engulfed pathogens [159]. The maturation block caused by LAMP deficiency is at least partially due to the inability of autophagosomes and phagosomes to undergo dynein/dynactin-mediated centripetal movement along microtubules towards lysosomes [158]. Interestingly LAMP-2 single knockout mice show an impaired phagosomal maturation in neutrophilic leucocytes. The impairment of this innate immune defense mechanism leads to periodontitis, which is one of the most widespread infectious diseases worldwide. The retarded clearance of bacterial pathogens is due to an inefficient fusion between lysosomes and phagosomes, leading to less efficient killing of the ingested pathogens [160], [161]. Neutrophils of the LAMP-2 knockout mice also contain an accumulation of autophagic vacuoles [85], [160], which is likely also due to impaired fusion of autophagosomes with lysosomes.
Taken together these observations indicate that fusion with lysosomes is required to successfully complete both autophagosome and phagosome maturation that is necessary for efficient degradation of the cargo. Further, the results show that the maturation of autophagosomes and phagosomes share common features, because both processes are impaired with similar tissue and cell specificity in LAMP-2 deficient mice and LAMP-double deficient cell lines.
7. Conclusions
Degradation of cytosolic proteins in lysosomes via autophagy has turned out to have numerous, partly unexpected, roles in health and disease. Autophagy has been shown to contribute to innate and adaptive immunity and longevity, and to the prevention of cancer and neurodegeneration, just to mention a few of its newly-revealed functions. Treatments for human diseases that specifically target autophagy do not yet exist. It is likely, however, that such treatments will emerge in the future, once the molecular mechanisms of the processes involved in autophagy regulation have been clarified and suitable inducers and inhibitors for clinical trials have been identified.
Full Paper : https://www.sciencedirect.com/science/article/pii/S0167488908002632