Authors – Adams LS, Kanaya N, Phung S, Liu Z, Chen S.
Abstract
Previous studies in our laboratory demonstrated that blueberry (BB) extract exhibited antitumor activity against MDA-MB-231 triple negative breast cancer (TNBC) cells and decreased metastatic potential in vitro.
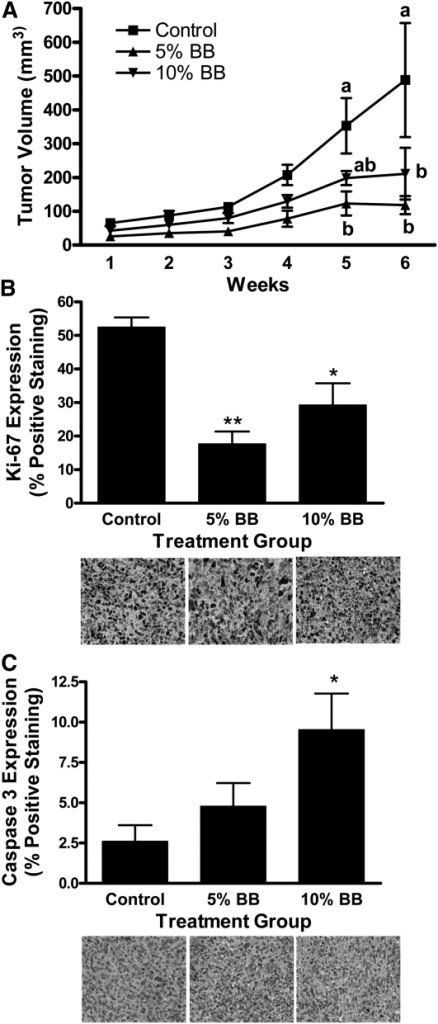
The current study tested 2 doses of whole BB powder, 5 and 10% (wt:wt) in the diet, against MDA-MB-231 tumor growth in female nude mice. In this study, tumor volume was 75% lower in mice fed the 5% BB diet and 60% lower in mice fed the 10% BB diet than in control mice (P ≤ 0.05).
Tumor cell proliferation (Ki-67) was lower in the 5 and 10% BB-fed mice and cell death (Caspase 3) was greater in the 10% BB-fed mice compared to control mice (P ≤ 0.05).
Gene analysis of tumor tissues from the 5% BB-fed mice revealed significantly altered expression of genes important to inflammation, cancer, and metastasis, specifically, Wnt signaling, thrombospondin-2, IL-13, and IFNγ. To confirm effects on Wnt signaling, analysis of tumor tissues from 5% BB-fed mice revealed lower β-catenin expression and glycogen synthase kinase-3β phosphorylation with greater expression of the β-catenin inhibitory protein adenomatous polyposis coli compared to controls.
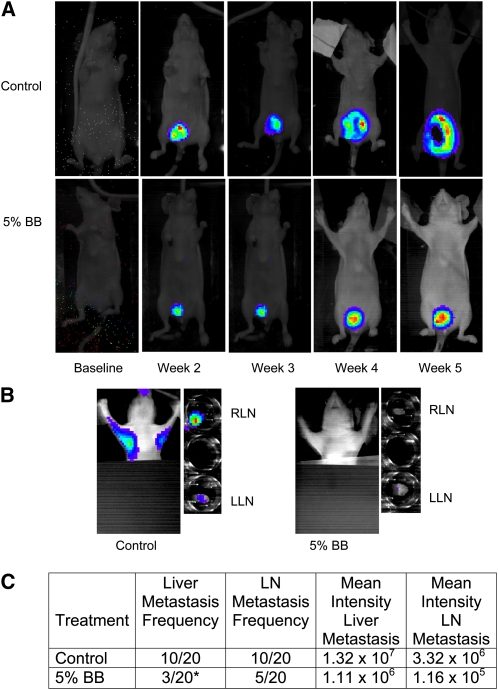
A second study tested the ability of the 5% BB diet to inhibit MDA-MB-231-luc-D3H2LN metastasis in vivo. In this study, 5% BB-fed mice developed 70% fewer liver metastases (P = 0.04) and 25% fewer lymph node metastases (P = 0.09) compared to control mice.
This study demonstrates the oral antitumor and metastasis activity of whole BB powder against TNBC in mice.
MDA-MB-231 tumor volume (A), proliferation (B), and apoptosis (C) in female nude mice fed control, 5% BB powder, or 10% BB powder diet for 8 wk. Data are means ± SEM, n = 6. In A, labeled means at a time without a common letter differ, P ≤ 0.05. In B and C, asterisks indicate different from control: *P ≤ 0.05, **P ≤ 0.01. BB, blueberry. In situ tumor growth and metastasis monitored by Xenogen IVIS imaging at different time points after MDA-MB-231 tumor implantation in female nude mice fed control or 5% BB diet for 7 wk. (A) Orthotopic breast tumor growth from baseline to wk 5 postimplantation. (B) In situ and ex-vivo imaging of RLN and LLN lymph node metastasis at wk 5 postimplantation. (C) Frequency of metastasis and mean intensity of liver and LN metastases analyzed by contingency table. Intensity data are mean ± SEM for the number of mice with metastases. *Different from control, P ≤ 0.05. BB, blueberry; LLN, left lymph node; RLN, right lymph node.
Particles of high energy and charge (HZE particles), which are abundant outside the magnetic field of the Earth, have been shown to disrupt the functioning of neuronal communication in critical regions of the brain. Previous studies with HZE particles, have shown that irradiation produces enhanced indices of oxidative stress and inflammation as well as altered neuronal function that are similar to those seen in aging.
Feeding animals antioxidant-rich berry diets, specifically blueberries and strawberries, countered the deleterious effects of irradiation by reducing oxidative stress and inflammation, thereby improving neuronal signaling. In the current study, we examined the effects of exposure to (56)Fe particles in critical regions of brain involved in cognitive function, both 36h and 30 days post irradiation.
We also studied the effects of antioxidant-rich berry diets, specifically a 2% blueberry or strawberry diet, fed for 8 weeks prior to radiation as well as 30 days post irradiation. (56)Fe exposure caused significant differential, neurochemical changes in critical regions of the brain, such as hippocampus, striatum, frontal cortex, and cerebellum, through increased inflammation, and increased oxidative stress protein markers. (56)Fe exposure altered the autophagy markers, and antioxidant-rich berry diets significantly reduced the accumulation of p62 in hippocampus, a scaffold protein that co-localizes with ubiquitinated protein at the 30 days post irradiation time-point.
Exposure to (56)Fe particles increased the accumulation of disease-related proteins such as PHF-tau in the hippocampus of animals fed the control diet, but not in the irradiated animals fed the blueberry diet. These results indicate the potential protective effects of antioxidant-rich berry diets on neuronal functioning following exposure to HZE particles.
Authors – Sangeetha Thangaswamy, Eric A. Bridenbaugh and Anatoliy A. Gashev
Abstract
Background
We have previously shown that aging is associated with weakened rat mesenteric lymphatic vessel (MLV) contractility. However, the specific mechanisms contributing to this aging-associated contractile degeneration remain unknown. Aging is often associated with elevations in oxidative stress, and reactive oxygen species (ROS) have been shown to reduce the contractility of MLV. Thus in the present study, we sought to assess whether aging is associated with increased levels of oxidative stress and oxidative damage in MLV.
Methods and Results
MLV were isolated from 9-mo- and 24-mo-old Fischer-344 rats and subjected to the following experimental techniques: measurement of total superoxide dismutase (SOD) activity; estimation of lipid peroxidation levels via measurement of thiobarbituric acid reactive substances (TBARS); detection of superoxide and mitochondrial ROS in live MLV; Western blot analysis, and immunohistochemical labeling of the SOD isoforms and nitro-tyrosine proteins. We found that aging is associated with increased levels of cellular superoxide and mitochondrial ROS concomitant with a reduction in Cu/Zn-SOD protein expression and total SOD enzymatic activity in MLV. This increase in oxidative stress and decrease in antioxidant activity was associated with evidence of increased lipid (as indicated by TBARS) and protein (as indicated by nitro-tyrosine labeling) oxidative damage.
Conclusions
Thus for the first time, we demonstrate that aging-associated increases in oxidative stress and oxidative damage is indeed present in the walls of MLV and may contribute to the aging-associated lymphatic pump dysfunction we previously reported.
Authors – Gaétan Chevalier – Stephen T. Sinatra – James L. Oschman – Karol Sokal – Pawel Sokal
Abstract
Environmental medicine generally addresses environmental factors with a negative impact on human health. However, emerging scientific research has revealed a surprisingly positive and overlooked environmental factor on health: direct physical contact with the vast supply of electrons on the surface of the Earth. Modern lifestyle separates humans from such contact.
The research suggests that this disconnect may be a major contributor to physiological dysfunction and unwellness. Reconnection with the Earth’s electrons has been found to promote intriguing physiological changes and subjective reports of well-being. Earthing (or grounding) refers to the discovery of benefits—including better sleep and reduced pain—from walking barefoot outside or sitting, working, or sleeping indoors connected to conductive systems that transfer the Earth’s electrons from the ground into the body.
This paper reviews the earthing research and the potential of earthing as a simple and easily accessed global modality of significant clinical importance.
Conclusion
De Flora et al. wrote the following: “Since the late 20th century, chronic degenerative diseases have overcome infectious disease as the major causes of death in the 21st century, so an increase in human longevity will depend on finding an intervention that inhibits the development of these diseases and slows their progress” [33].
Could such an intervention be located right beneath our feet? Earthing research, observations, and related theories raise an intriguing possibility about the Earth’s surface electrons as an untapped health resource—the Earth as a “global treatment table.” Emerging evidence shows that contact with the Earth—whether being outside barefoot or indoors connected to grounded conductive systems—may be a simple, natural, and yet profoundly effective environmental strategy against chronic stress, ANS dysfunction, inflammation, pain, poor sleep, disturbed HRV, hypercoagulable blood, and many common health disorders, including cardiovascular disease. The research done to date supports the concept that grounding or earthing the human body may be an essential element in the health equation along with sunshine, clean air and water, nutritious food, and physical activity.
Authors : C. Linhart · D. Davidson · S. Pathmanathan · T. Kamaladas · C. Exley
Abstract:
Human exposure to aluminium is a burgeoning issue. The brain is a sink for systemically available aluminium and a putative target of neurotoxicity. An increasing number of studies continue to confirm the presence of aluminium in human brain tissue though primarily in relation to donors who have died of a neurodegenerative or neurodevelopmental disorder.
Herein, we have measured aluminium in brain tissue in donors who died of a specific disease or condition though without showing any neurodegeneration. The donors were diagnosed as not suffering from multiple sclerosis. Herein, these novel data are compared with recent data on aluminium in brain tissue in multiple sclerosis. Brain tissues from all four lobes were obtained from the Multiple Sclerosis Society Tissue Bank.
Tissues were digested using microwave-assisted acid digestion and their aluminium content was measured by transversely heated graphite furnace atomic absorption spectrometry. Both are established methods in our laboratory. Detailed statistical analyses were used to compare new data with recent data for multiple sclerosis.
Aluminium was found in brain tissue in each donor with a high proportion of measurements (189/291) being below 1.00 μg/g dry weight. The data for all cases (median and IQR) were 0.74 (0.48–1.28), 1.23 (0.62–1.63), 0.84 (0.45–1.14) and 1.01 (0.62–1.65) μg/g dry weight for occipital, parietal, temporal and frontal lobes, respectively.
There was a statistically significant positive correlation between aluminium content of brain tissue and the age of donor. Comparison of data for this non-multiple sclerosis group with brain aluminium data for donors dying with a diagnosis of multiple sclerosis showed that the latter had a statistically significant higher content of brain aluminium.
The data reinforce a previous conclusion that the aluminium content of brain tissue in multiple sclerosis is elevated and support the suggestion that human exposure to aluminium may have a role to play in the aetiology of multiple sclerosis.
Authors : Li Y, Wang J , Yue J , Wang Y, Yang C, Cui Q
Cell Biology International : 2018 February
Abstract
Magnesium, as a physiological calcium antagonist, plays a vital role in the bone metabolism and the balance between magnesium and calcium is crucial in bone physiology. We recently demonstrated that matrix mineralization in human bone marrow-derived mesenchymal stem cells (hBMSCs) can be suppressed by high Mg2+ .
However, a complete understanding of the mechanisms involved still remains to be elucidated. As mitochondrial calcium phosphate granules depletion manifests concurrently with the appearance of matrix vesicles (MVs) and autophagy are associated with matrix mineralization, we studied the effect of high extracellular Mg2+ on these pathways.
Our results first demonstrated that high Mg2+ has a significant inhibitory effect on the generalization of extracellular mineral aggregates and the expression of collagen 1 along which the mineral crystals grow. Transmission electron microscope results showed that less amount of MVs were observed inside hBMSCs treated with high Mg2+ and high Mg2+ inhibited the release of MVs.
In addition, high Mg2+ significantly suppressed mitochondrial Ca2+ accumulation. Autophagy is promoted as a response to osteogenesis of hBMSCs. High Mg2+ inhibited the level of autophagy upon osteogenesis and autophagy inhibitor 3-MA significantly suppressed mineralization. Exogenous ATP can reverse the inhibitory effect of high Mg2+ by increasing the level of autophagy.
Taken together, our results indicate that high Mg2+ may modulate MVs-mediated mineralization via suppressing mitochondrial Ca2+ intensity and regulates autophagy of hBMSCs upon osteogenesis, resulting in decreased extracellular mineralized matrix deposition. Our results contribute to the understanding of the role of magnesium homeostasis in osteoporosis and the design of magnesium alloys.
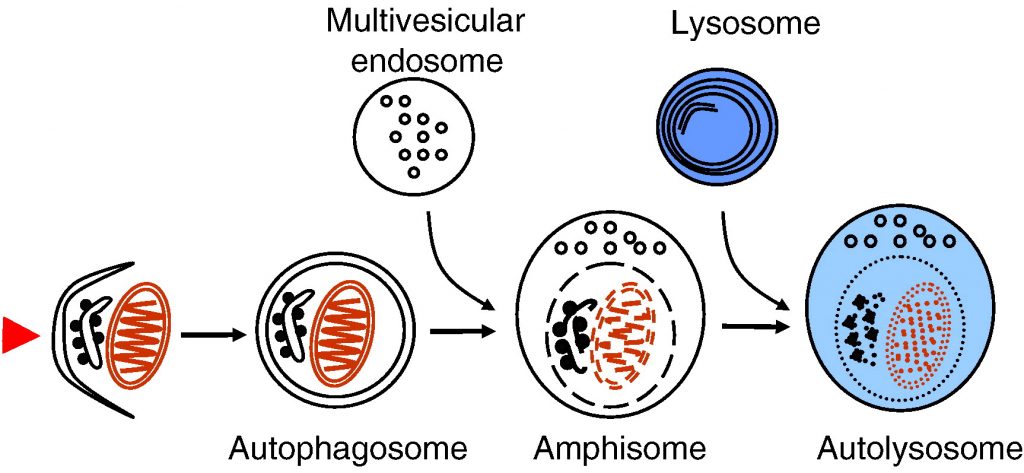
Autophagy delivers cytoplasmic material and organelles to lysosomes for degradation. The formation of autophagosomes is controlled by a specific set of autophagy genes called atg genes. The magnitude of autophagosome formation is tightly regulated by intracellular and extracellular amino acid concentrations and ATP levels via signaling pathways that include the nutrient sensing kinase TOR. Autophagy functions as a stress response that is upregulated by starvation, oxidative stress, or other harmful conditions. Remarkably, autophagy has been shown to possess important housekeeping and quality control functions that contribute to health and longevity. Autophagy plays a role in innate and adaptive immunity, programmed cell death, as well as prevention of cancer, neurodegeneration and aging. In addition, impaired autophagic degradation contributes to the pathogenesis of several human diseases including lysosomal storage disorders and muscle diseases.
Fig. 1. Schematic presentation of autophagosome formation and maturation by fusion with endosomes and lysosomes. The arrowhead on the left illustrates the induction signal that initiates the process.
5. Physiological functions of autophagy
5.1. Stress response and housekeeping function
The role of autophagy as a survival mechanism during short-term amino acid starvation is well documented. Macroautophagy is induced by starvation of serum and amino acids; autophagosomes can be detected already after 15–30 min of starvation [55]. During long-term starvation, chaperone-mediated autophagy increases and macroautophagy decreases [80], [81]. Yeast strains defective in autophagy do not survive nitrogen starvation [38]. Knockout mice deficient in one of the central autophagy proteins, Atg5, show that autophagy is indispensable for the energy metabolism immediately after birth [82]. Atg5 knockout mice die of starvation one day after birth.
In muscle and heart cells, autophagy seems to have a special housekeeping role in the turnover of cytoplasmic constituents including mitochondria. This is revealed by myopathy and cardiomyopathy in patients and mice possessing a defective autophagic degradation due to deficiency of the lysosomal membrane protein LAMP-2 [83], [84], [85]. LAMP-2 deficiency is described in detail later in this review. The importance of autophagy for the heart muscle is supported by a study showing that heart-specific loss of the autophagy protein Atg5 causes cardiomyopathy in mice [86]. Evidence has been published suggesting that damaged mitochondria might be autophagocytosed selectively in a process termed mitophagy [87]. Mitochondria are the major source of reactive oxygen species in cells. Interestingly, reactive oxygen species are necessary for the signal transduction pathway initiating starvation-induced autophagy [88].
It was proposed long ago that autophagy has a role in growth regulation, as suggested by decreased autophagy during growth of the kidney after unilateral nephrectomy [89]. Inducible knockdown of the autophagy protein Atg5 in cell culture shows that autophagy negatively controls cell size [90]. Similar result was observed in Drosophila fat body over-expressing the autophagy protein Atg1 [91].
Autophagy contributes to intracellular quality control and housekeeping, especially in turnover of aggregate-prone proteins. Prevention of autophagy by conditional knockout of atg7 leads to accumulation of ubiquitinated protein aggregates in mouse tissues [92]. Tissue-specific knockout of autophagy proteins in the central nervous system causes accumulation of ubiquitin-positive protein aggregates and neurodegeneration in mice [93], [94]. Further, enhanced autophagy reduces the toxicity of the Huntingtin protein aggregates that accumulate in Huntington disease [95]. Autophagy may prevent aggregate formation by degrading the proteins as monomers, oligomers, or after aggregate formation [96]. It is not clear at present whether aggregated proteins are segregated preferentially, or whether they are removed via unspecific autophagic uptake of cytoplasm. Two proteins have been proposed to function during the uptake of protein aggregates: Alfy and p62 [97], [98]. p62 binds to both ubiquitin-conjugated aggregate-prone proteins and the autophagosome protein LC3 [99], which suggests that it could selectively recruit autophagy machinery to the aggregates and enhance their autophagic clearance.
In addition to removal of cytoplasmic aggregate-prone proteins, autophagy also contributes to the quality control in the ER. Unfolded protein response induces autophagy, and this induction is beneficial for cell survival [100], [101], [102].
Autophagic degradation is also needed for early embryonic development. A recent study shows that autophagy-defective mouse eggs fertilized with autophagy-defective sperm, failed to develop beyond the four and eight cell stages [103]. The authors suggest that autophagy may be needed in the preimplantation embryos for protein recycling, production of amino acids for protein synthesis or substrates for energy production, or for removal of obsolete maternal factors.
5.2. Innate and adaptive immunity
Autophagy contributes to both innate and adaptive immunity [104], [105]. In some cases autophagy can protect cells against intracellular pathogens. Induction of autophagy during Herpes simplex virus infection, and localization of viral particles inside autophagic vacuoles, were proposed to indicate that autophagy acts as a host-defense mechanism in infected cells [59]. The Herpes virus virulence protein, ICP34.5, inhibits autophagy, suggesting that the virus has developed a way to prevent the autophagic defense of the host cell. Autophagy may also help cells to defend against intracellular bacteria [106]. Sequestration of intracellular Group A Streptococci in autophagosome-like structures protects cells against the bacteria [107]. Mycobacterium tuberculosis is normally able to survive inside macrophages by preventing the fusion of phagosomes with lysosomes. Surprisingly, induction of autophagy bypasses the maturation defect, leading to phagolysosome formation and bacterial killing [108].
Macroautophagy also contributes to antigen presentation. Major histocompatibility complex (MHC) class II molecules present products of lysosomal proteolysis to CD4(+) T cells. Extracellular antigen uptake is considered to be the main source of MHC class II ligands. However, it was demonstrated that in MHC class II-positive cells, including dendritic cells, B cells, and epithelial cells, autophagosomes continuously fuse with multivesicular MHC class II-loading compartments [109]. This pathway is of functional relevance, because targeting of the influenza matrix protein 1 to autophagosomes enhances its MHC class II presentation to CD4(+) T cells. Thus it seems that macroautophagy efficiently delivers cytosolic proteins for MHC class II presentation and can improve helper T cell stimulation.
5.3. Cell death
Autophagy also seems to have roles in programmed cell death [110], [111]. Type II programmed cell death, or autophagic cell death, was originally described in mammary carcinoma cells [112], [113]. Autophagy proteins were shown to be necessary for cell death under certain conditions, such as in apoptosis-defective cells [114], [115], [116]. In this scenario autophagy is needed for the execution of cell death. Under other conditions, such as nutrient starvation, autophagy protects cells against apoptosis by providing nutrients [117], [118], [119]. The regulation of apoptosis and autophagy are linked via the antiapoptotic protein Bcl-2. Bcl-2 inhibits Beclin 1-dependent autophagy by binding to Beclin 1 and preventing its association with Vps34 [52]. This anti-autophagy function of Bcl-2 was proposed to help maintain autophagy at levels that are compatible with cell survival, rather than cell death.
Lipids may also regulate autophagy and its outcome to the host cell. Ceramide and sphingosine 1-phosphate, a metabolite of ceramide, both induce autophagy in mammalian cells [120]. The outcome on cell survival is however different: ceramide promotes cell death, while sphingosine 1-phosphate increases cell survival. Ceramide is part of the signaling cascade initiated by chemotherapy, while sphingosine 1-phosphate is part of the signaling cascade initiated by starvation. Beclin 1 level and the autophagy response are stronger during ceramide signaling [121].
Autophagy has functions in cell death during development [122]. atg genes are necessary for the clearance of apoptotic cells during embryonic development in mice [123]. Autophagy contributes to dead-cell clearance during programmed cell death by maintaining cellular energy levels in the dying cells, thereby allowing the generation of cell surface and secreted signals that then promote engulfment of cell corpses by neighboring cells. Autophagy is also indispensable for the execution of certain types of cell death during development. The degradation of Drosophila salivary glands by type II programmed cell death depends on autophagy [73].
5.4. Aging and longevity
Finally, autophagy also contributes to longevity [124]. Reduced caloric intake increases longevity in several animal species. Increased autophagic turnover of cytoplasmic constituents including mitochondria was shown to contribute to the longer life in the dieting animals [125]. Further evidence that autophagy contributes to longevity come from Caenorhabditis elegans mutants possessing a defective insulin receptor (daf2 mutant), which live longer than control worms. The increased lifetime of these mutant worms depends on a functional autophagic pathway [126]. Moreover, knockdown of autophagy gene products including Atg7 and Atg12 were shown to shorten the lifespan of both wild type and daf2 mutant C. elegans[127]. Further, promoting basal levels of autophagy in the nervous system of adult Drosophila enhances longevity of the flies [128]. Together these studies give strong support for a role of autophagy in the prevention of aging.
Fig. 6 summarizes the physiological functions of autophagy described above.
Fig. 6. A summary of the functions of autophagy in health and disease.
6. Autophagy and disease
6.1. Cancer
Impaired autophagy contributes to cancer development [2], [129], [130]. Beclin 1 is monoallelically deleted in a large proportion of human breast and ovarian cancers. Over-expression of Beclin 1 in a breast cancer cell line increases autophagy and decreases the growth and tumorigenicity of these cells [131]. Mice with heterozygous deletion of Beclin 1 have less autophagy and more tumors than control mice [132], [133]. Further, the other autophagy-promoting components of the Beclin 1/Vps34 complex, UVRAG and Ambra 1 (Fig. 4), are also tumor suppressors [48], [49]. Moreover, knockout of Bif-1, also part of the Beclin 1 complex, significantly enhances the development of spontaneous tumors in mice [50]. On the other hand, binding of the proto-oncogenic proteins Bcl-2 or Bcl-XL to Beclin 1 inhibit autophagy [51], [52]. In addition to the Beclin 1 complex, other tumor suppressors also enhance autophagy. PTEN is a phosphatase that decreases the concentration of class I PI3 kinase product and enhances autophagy [72]. PTEN is also a tumor suppressor [134]. Further, the activities of Ras and class I PI3-kinases inhibit autophagy and promote cell growth. Ras is mutated and class I PI3 kinases are upregulated in many cancers [135], [136].
The results described above show that autophagy contributes to the prevention of tumorigenesis. Impaired autophagy can contribute to tumor formation via impaired regulation of cell growth, and/or via decreased cell death. In addition, it was shown that failure to sustain metabolism via autophagy results in increased DNA damage. This chromosomal instability increases tumor progression [137].
In advanced cancers, autophagy may have the opposite effect on the tumor development. Autophagy can benefit the progression of the tumor because it can provide nutrients during starvation [129], [130], [138]. In addition, autophagy was recently shown to improve the survival of p53-deficient cancer cells under starvation or hypoxic conditions [139]. These findings suggest that autophagy inhibition, rather than stimulation, might be beneficial in treatment of advanced cancer.
6.2. Neurodegeneration
Many age-related neurodegenerative diseases are characterized by the accumulation of ubiquitin-positive protein aggregates in affected brain regions. These misfolded, aberrant proteins can disrupt neuronal function and cause neurodegeneration. As described earlier, autophagy is necessary for the clearance of aggregate-prone proteins that are toxic especially for post-mitotic cells like neurons [130]. Tissue-specific knockout of the autophagy genes in neurons causes a massive accumulation of ubiquitin-positive protein aggregates and neurodegeneration in mice [93], [94], indicating that autophagy is needed for the constitutive clearance of aggregate-prone proteins. Autophagy was recently shown to enhance the clearance of Huntingtin, mutant tau, synphilin 1 and α-synuclein, but not AIMP2 (p38) and mutant desmin [140]. This study indicates that autophagy is not able to degrade all protein aggregates. However, the role of autophagy has been demonstrated in Huntington’s disease, caused by mutations in Huntingtin, and familial Parkinson’s disease, caused by mutations in α-synuclein. Enhanced autophagy in animal models of these diseases improves clearance of the aggregated proteins and reduces the symptoms of neurodegeneration [95], [141].
ESCRT complexes are necessary for the biogenesis of multivesicular endosomes. As described earlier, multivesicular endosomes are necessary for the maturation of autophagosomes into degradative autolysosomes. Mutations in ESCRT III subunits CHMP2B or mSnf7-2 are associated with two neurodegenerative diseases, frontotemporal dementia and amyotropic lateral sclerosis. Both diseases are characterized by abnormal ubiquitin-positive protein deposits in affected neurons. Cell lines and fruit flies depleted of CHMP2B or mSnf7-2 show decreased autophagic degradation, increased levels of ubiquitin-positive aggregates and increased neurodegeneration [27], [29].
Alzheimer’s disease is characterized by the accumulation of extracellular amyloid plaques in the brain. These plaques consist of aggregated β-amyloid (Aβ) peptide. Autophagy was proposed to contribute to the production of Aβ. Autophagic compartments containing both amyloid precursor protein and Aβ accumulate in dystrophic neurons in Alzheimer brain [142], [143]. Purified autophagic vacuoles contain all necessary constituents for Aβ production [142], and autophagic compartments were identified as a major reservoir of intracellular Aβ in the brain of Alzheimer patients and mouse models. The primary cause for the increased accumulation of autophagic compartments in Alzheimer’s disease was recently suggested to be their retarded maturation to autolysosomes [144].
A recent study, however, challenges the idea that autophagy contributes to the pathogenesis of Alzheimer’s disease. Beclin 1 was shown to be decreased in affected brain regions of patients with Alzheimer disease early in the disease process [145]. Heterozygous deletion of Beclin 1 in mice decreased neuronal autophagy and resulted in neurodegeneration. Transgenic mice expressing human amyloid precursor protein have been used as a mouse model for this disease. Genetic reduction of Beclin 1 expression increased intraneuronal Aβ accumulation, extracellular Aβ deposition, and neurodegeneration [145]. Increasing Beclin 1 levels by lentiviral expression reduced both intracellular and extracellular amyloid pathology in these transgenic mice. This study suggests that decreased, not increased, autophagy promotes Alzheimer’s disease progression. Further, enhancing autophagy by increasing Beclin 1 levels may have therapeutic potential in this disease.
6.3. Autophagy and lysosomal storage diseases
Niemann–Pick type C is a neurodegenerative lipid storage disorder characterized by a disruption of sphingolipid and cholesterol trafficking caused by mutations in either of two genes, npc1 and npc2. The disease produces cognitive impairment, ataxia and death, often in childhood. Cells deficient in npc genes show increased expression of Beclin 1 and LC3-II, the autophagosome-specific form of LC3, suggesting autophagy is induced [146]. Increased levels of LC3-II have also been observed in npc-deficient brain tissue [147]. npc-Deficient cerebellar Purkinje neurons undergo a cell death that was proposed to depend on autophagy [148], suggesting increased autophagy may be harmful for neurons in NPC patients.
Most lysosomal storage diseases are caused by deficiencies of lysosomal hydrolases, leading to accumulation of undegraded substrate and other material in the lysosomal compartment. Lysosomal accumulation of substrates can also affect autophagosome–lysosome fusion. Autophagosomes accumulate in brain and isolated cell lines of mouse models of two lysosomal storage diseases associated with severe neurodegeneration, multiple sulfatase deficiency and mucopolysaccharidosis type IIIA [149]. Significantly reduced colocalization of the lysosomal membrane protein LAMP-1 with the autophagosome marker LC3 indicates that the fusion of lysosomal compartments with autophagosomes is impaired. In addition, cell lines isolated from these mice have decreased ability to degrade aggregate-prone proteins and show accumulation of polyubiquitinated proteins and non-functional mitochondria. Thus, neurodegeneration observed in many lysosomal storage diseases may be at least partially due to impaired autophagic degradation, which is particularly vital for neurons.
6.4. Autophagy and muscle disorders
Autophagic vacuoles are a frequent feature in numerous muscular disorders. Such a pathological situation can be observed in patients suffering from Danon disease, an inherited disease resulting from null mutations in the lysosomal membrane protein LAMP-2 [83]. LAMP-2 deficiency leads to a fatal cardiomyopathy and myopathy sometimes associated with mental retardation [150]. Accumulation of autophagic vacuoles in the heart and skeletal muscle are hallmarks of the disease [84]. Studies in LAMP-2 deficient mice revealed in part similar findings [85]. Fifty percent of these mice die at an early postnatal age with massive accumulation of autophagic vacuoles in several tissues including liver, pancreas, spleen, kidney, lymph nodes, neutrophilic leukocytes, skeletal muscle, and heart. Autophagic vacuoles containing single mitochondria were frequently observed in cardiomyocytes [151] indicating that mitochondria are a main target for autophagic degradation in muscle tissues. These cellular alterations lead to a reduced contractility and an increased size of the heart in LAMP-2 knockout mice. This is in agreement with the finding that cardiomyopathy is the hallmark in Danon disease patients [83]. Biochemical and electron microscopy studies reveal that a retarded consumption, rather than increased formation, of autophagic vacuoles leads to their accumulation [152]. LAMP-double deficient fibroblasts lack both LAMP-2 and the structurally related LAMP-1 protein. These cells show a defect in the final maturation steps of late autophagic vacuoles, involving retarded fusion with lysosomes [33], [119]. Interestingly, recruitment of the small GTPase Rab7 to autophagosomes is retarded in these cells [32].
Inhibition of lysosomal fusion using hydroxy-chloroquine causes similar vacuolar alterations and myopathies to Danon disease, confirming the important role of lysosome–autophagosome fusion for muscle cell physiology [153]. Impaired autophagosome maturation may also be related to other types of diseases such as X-linked myopathy with excessive autophagy, infantile autophagic vacuolar myopathy, adult-onset autophagic vacuolar myopathy with multiorgan involvement, and X-linked congenital autophagic vacuolar myopathy [154]. The molecular defects in these disorders are still unknown.
Although altered autophagy has been observed in various heart diseases, including cardiac hypertrophy and heart failure, it remains unclear whether autophagy plays a beneficial or detrimental role in these diseases. As mentioned earlier, tissue-specific deletion of atg5 in heart causes cardiac hypertrophy and contractile dysfunction [86]. In addition, increased levels of ubiquitinated proteins and abnormal mitochondria are found, especially after treatment with pressure overload or β-adrenergic stress. This suggests that autophagy is needed in the heart to ensure the availability of sufficient energy substrates and to control cardiomyocyte size and global cardiac structure and function.
6.5. Common aspects in autophagy and phagocytosis
As described above, autophagy plays a role in innate immunity against intracellular pathogens [104], [105] by clearing microbes directly via ingestion into autophagosomes for subsequent degradation in autolysosomes [108], [155]. Similar to the process of intracellular defense, phagocytosis is an evolutionary conserved mechanism involved in the removal of extracellular organisms. Interestingly, it was found that the phagocytic and autophagic pathways are linked. Toll like receptor (TLR) activation triggers the recruitment of autophagy proteins LC3, Atg5 and Atg7 to the phagosomal pathway. Before these events Beclin 1 and class III PI3-kinase activity are found in phagosomes [156]. These autophagy-specific proteins are recruited to the phagosome, while almost no classical autophagosomes are observed in the cells. Phagosome fusion with lysosomes is then initiated, leading to acidification and killing of the ingested organisms. Thus, engaging the autophagy pathway via TLR signaling (especially TLR7 through its binding to single-stranded RNA) enhances phagosome maturation and destruction of pathogens [157]. This underscores the intimate link between autophagy and phagocytosis.
As mentioned earlier, M. tuberculosis is able to survive inside macrophages by preventing the fusion of phagosomes with lysosomes, but induction of autophagy bypasses the maturation defect, leading to phagolysosome formation and bacterial killing [108]. Autophagy induction induces the localization of Beclin 1 and LC3 to phagosomes, suggesting the phagosomes are diverted to an autophagosome-like compartment that is then able to fuse with lysosomes.
The association between autophagy and phagocytosis is also underlined by studies with cells lacking either one or both LAMPs [33]. As described above, autophagosome–lysosome fusion is impaired in LAMP-double deficient cells [32]. Whereas macrophages and fibroblasts from LAMP-1 or LAMP-2 single-deficient mice display normal fusion of lysosomes with phagosomes, in LAMP-double knockout fibroblasts phagosomes are unable to recruit late endosomal/lysosomal markers and phagocytosis is arrested prior to the acquisition of Rab7 [158]. Interestingly, the maturation of Neisseria-containing phagosomes is also disturbed and cells lacking both LAMP proteins fail to kill the engulfed pathogens [159]. The maturation block caused by LAMP deficiency is at least partially due to the inability of autophagosomes and phagosomes to undergo dynein/dynactin-mediated centripetal movement along microtubules towards lysosomes [158]. Interestingly LAMP-2 single knockout mice show an impaired phagosomal maturation in neutrophilic leucocytes. The impairment of this innate immune defense mechanism leads to periodontitis, which is one of the most widespread infectious diseases worldwide. The retarded clearance of bacterial pathogens is due to an inefficient fusion between lysosomes and phagosomes, leading to less efficient killing of the ingested pathogens [160], [161]. Neutrophils of the LAMP-2 knockout mice also contain an accumulation of autophagic vacuoles [85], [160], which is likely also due to impaired fusion of autophagosomes with lysosomes.
Taken together these observations indicate that fusion with lysosomes is required to successfully complete both autophagosome and phagosome maturation that is necessary for efficient degradation of the cargo. Further, the results show that the maturation of autophagosomes and phagosomes share common features, because both processes are impaired with similar tissue and cell specificity in LAMP-2 deficient mice and LAMP-double deficient cell lines.
7. Conclusions
Degradation of cytosolic proteins in lysosomes via autophagy has turned out to have numerous, partly unexpected, roles in health and disease. Autophagy has been shown to contribute to innate and adaptive immunity and longevity, and to the prevention of cancer and neurodegeneration, just to mention a few of its newly-revealed functions. Treatments for human diseases that specifically target autophagy do not yet exist. It is likely, however, that such treatments will emerge in the future, once the molecular mechanisms of the processes involved in autophagy regulation have been clarified and suitable inducers and inhibitors for clinical trials have been identified.
Authors : Yong-Bo Hu Eric B Dammer Ru-Jing Ren Gang Wang
Abstract
The endosomal-lysosomal system is made up of a set of intracellular membranous compartments that dynamically interconvert, which is comprised of early endosomes, recycling endosomes, late endosomes, and the lysosome. In addition, autophagosomes execute autophagy, which delivers intracellular contents to the lysosome. Maturation of endosomes and/or autophagosomes into a lysosome creates an unique acidic environment within the cell for proteolysis and recycling of unneeded cellular components into usable amino acids and other biomolecular building blocks.
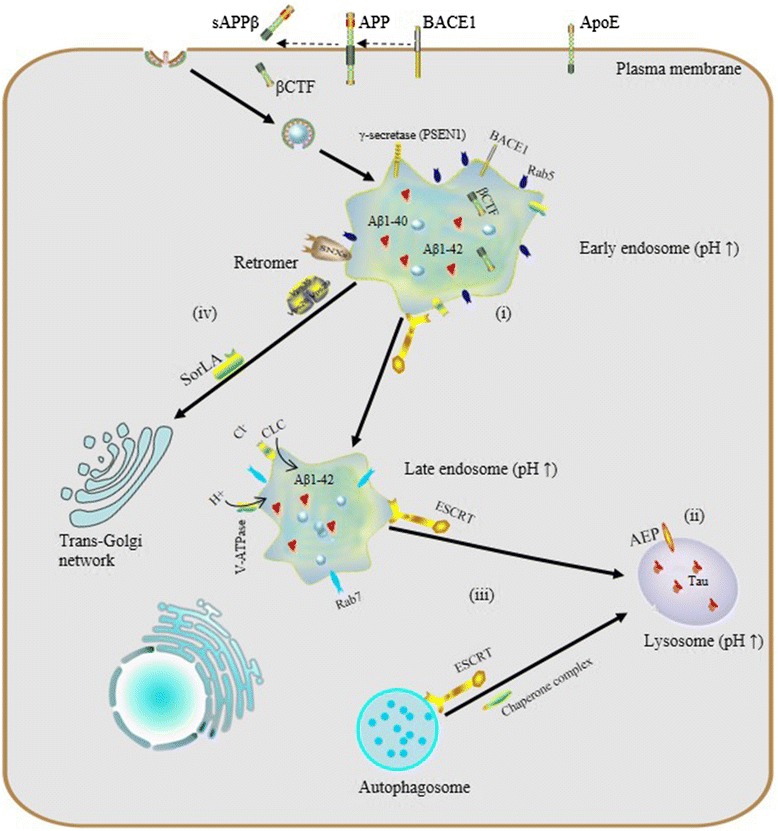
In the endocytic pathway, gradual maturation of endosomes into a lysosome and acidification of the late endosome are accompanied by vesicle trafficking, protein sorting and targeted degradation of some sorted cargo. Two opposing sorting systems are operating in these processes: the endosomal sorting complex required for transport (ESCRT) supports targeted degradation and the retromer supports retrograde retrieval of certain cargo.
The endosomal-lysosomal system is emerging as a central player in a host of neurodegenerative diseases, demonstrating potential roles which are likely to be revealed in pathogenesis and for viable therapeutic strategies. Here we focus on the physiological process of endosomal-lysosomal maturation, acidification and sorting systems along the endocytic pathway, and further discuss relationships between abnormalities in the endosomal-lysosomal system and neurodegenerative diseases, especially Alzheimer’s disease (AD).
The endosomal-lysosomal system is a series of organelles in the endocytic pathway where various cargo molecules required for normal cellular function are internalized, recycled and modulated. Recently, mounting evidence has suggested that abnormalities in both endosomes and lysosomes, or dysregulation in their trafficking, play an important role directly in a surprising host of neurological dysfunctions, represented by AD, Parkinson’s disease (PD), and Lewy body dementia (LBD) [1–3]. Thus, the endosomal-lysosomal system is emerging as a key to understanding the mechanisms underlying both protein degradation and neurodegeneration. Here, we intend to summarize advances in the study of the endosomal-lysosomal system, with a focus on compartmentalized organization of trafficking routes, sorting machinery and their relationships to neurodegeneration.
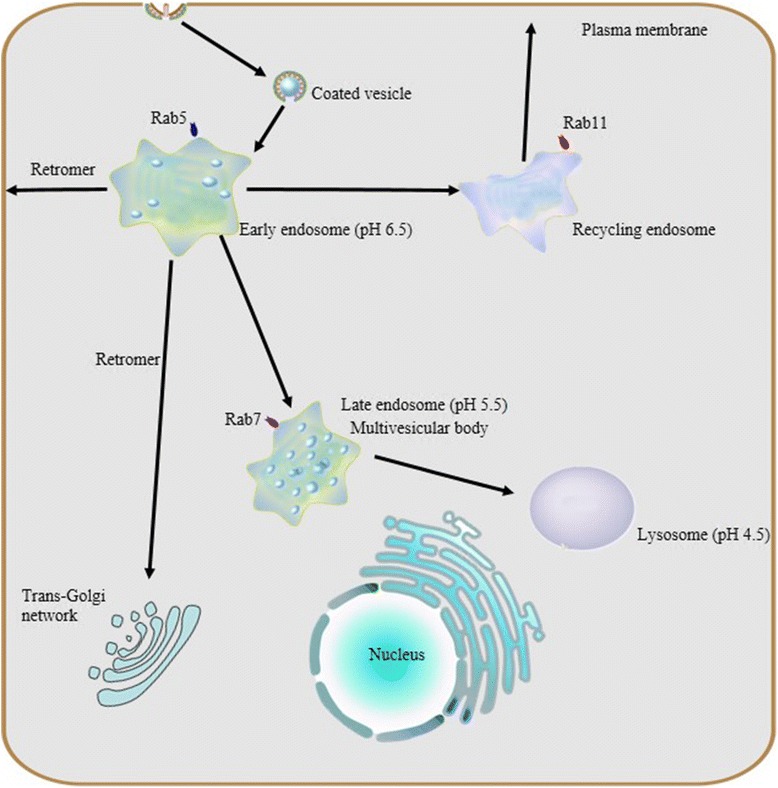
Endocytosis and endosome maturation. Protein internationalization is often dependent on a ubiquitous clathrin-mediated mechanism. Subsequent to internationalization, cargo proteins are transported to early endosomes via endocytic vesicles under the control of Rab5. Early endosomes serve as the major sorting stations where proteins can be sorted into recycling endosomes for recycling back to the cell surface, into a retrogradee pathway mediated by retromer to be sent back to the trans-golgi network (TGN), or into a degradation pathway for eventual targeting to the lysosome. As the number of intraluminal vesicles increases, early endosomes mature into late endosomes, and then late endosomes fuse with lysosomes.Due to their different capacities of acidification, a stable pH gradient is established in different compartments during the maturation process: early endosomes maintain pH at about 6.5, late endosomes at about 5.5 and lysosomes at about 4.5
A pH gradient established by vacuolar ATPase and chloride channels
Vacuolar ATP-dependent acidification
As mentioned above, the endosomal and lysosomal compartments share at least one similar significant characteristic: low intraluminal pH. These sealed acidic compartments provide an appropriate environment for optimal degradation of substrate cargo and recycling of their cognate receptors. As macromolecules are transported along the endosomal-lysosomal pathway, the internal pH of both endosomes and lysosomes decreases rapidly due to vacuolar acidification.
Previous research revealed that the same H+-ATPase, also known as vacuolar ATPase (V-ATPase), acidifies both endosomes and lysosomes. V-ATPase, differs from Na+,K+-ATPase in the plasma membrane, Ca2+-ATPase in the sarcoplasmic reticulum, and F1,F0-ATP synthase in mitochondria, in that it does not require a coupled influx of permeant anions [19]. Sulfhydryl alkylating reagents such as N-ethylmaleimide inhibit the V-ATPase dependent acidification of the endosomal-lysosomal system [19–21] as well as the specific inhibitor Bafilomycin A1 [22, 23].
V-ATPase is a unique class of ATPase present throughout the membranes which constrain the endocytic pathway, including the trans golgi network (TGN). V-ATPase, as a protein complex, is composed of two multimeric subunits, V1 in the cytoplasmic domain and V0 within the vacuolar membrane; the activity of V-ATPase depends on the dynamic assembly of these. V-ATPase is widely expressed in eukaryotic cells and serves as the master regulator of vesicular acidification in many subcellular membrane bound organelles. It also has important roles to play in vesicular trafficking and proteostasis.
Abnormalities and dysregulation of the endosomal-lysosomal system in neurodegeneration
Increasing attention being paid to the endosomal-lysosomal system has begun to elucidate a relationship between endosomal-lysosomal defects and neurodegeneration. In particular, robust pathology implicating endosomal-lysosomal disruption in AD has been well characterized. Here, we focus preferably on AD as a example of neurodegenerative disease and we believe that AD represents a general model of neurodegenerative diseases on abnormalities of the endosomal-lysosomal system occur along a continuum that includes early endosome changes, dysregulated acidification and sorting component defects.
The endosomal–lysosomal system and AD. (i) endosomal enlargement, Rab5 overexpression and Aß accumulation; (ii) dysregulated endosomal-lysosomal acidification, pH elevation and tau aggregation; (iii) dysfunctional ESCRT complexes, defective autophagy and accumulation of intraluminal ubiquitinated proteins; (iv) defects of retromer: reduction of Vps26, Vps35 and SorLA disrupts the trafficking and processing of APP
Dysregulated acidification, cellular indigestion?
Endosomal-lysosomal pH defects are an emerging theme in mechanisms underlying a number of neurodegenerative diseases. To date, results from experiments in vivo and in vitro have revealed the importance of proper vesicular pH balance and optimal acidification in transporting and degrading cargo via the endocytic pathway [48, 49]. For instance, Lee et al. reported that, in presenilin1 (PSEN1)-deleted blastocysts, defective lysosome acidification was observed with a substaintially elevated lysosomal PH of 5.4 and PSEN1 is essential for the transport of mature V0a1 subunites of V-ATP to lysosomes for their acidification and proteolysis [50].
Specifically, dysregulation of acidification and intracellular pH perturbation could influence the activity of enzymes in endomembrane compartments, resulting in impaired clearance of protein aggregates downstream of elevated endomembrane system pH, or conversely, due to decreased cytoplasmic pH. Regarding the latter, asparaginyl endopeptidase (AEP) is a typical pH-sensitive protein hydrolase the activity of which depends on the acidic pH of vesicular compartments. Predominantly localized in late endosomes, asparaginyl endopeptidase (AEP) specifically cleaves substrates with an asparagine residue at the P1 site. It is known that AEP can undergo reversible pH-dependent autoproteolytic activation, and in normal conditions, full-length pro-AEP is inactive [51].
As pH decreases from neutral to acidic, the activity of AEP gradually increases, such that it is partially activated at pH 4.5 and fully activated at pH 3.5, via removal of a cap that covers the active site. In AD patients, lysosomal acidification may be defective and it has been shown that the intracellular pH of neurons gradually decreases with aging [52] and more so with lactic acid elevation seen in AD cortex [53], so ectopic AEP activation or activity after leakage of active enzyme from late endosomes or lysosomes may be increased. AEP is involved in pathological tau degradation. Specifically, AEP generates tau fragments that form insoluble fibrils and result in neurotoxicity and neuropathological changes in AD [54, 55].
Increased endosome and lysosome pH is expected to have global effects on the proteome, particularly membrane proteins which rely on this pathway for their regulation and degradation. Interestingly, studies of microglia in culture have shown that in the absence of inflammatory IL-6 signaling, microglia do not achieve a sufficiently low lysosomal pH to degrade Aß, while after stimulation, CLC7 trafficking to lysosomes increases and pH drops sufficiently to improve Aß clearance [49, 56].
We have recently performed a systematic look at the proteomic effects of defective endosomal-lysosomal pH in a cellular model, in order to develop a better understanding of the global changes in the proteome that follow inhibition of V-ATPase and could be considered together as a signature or biomarker of defective vacuolar acidification [57, 58], which would be expected to have an overlap with changes seen in AD and/or other conditions which may be subject to this often age-dependent defect.
Indeed, blocking lysosomal degradation with bafilomycin A1 affects a significant increase in global K63 polyubiquitin linkages, which also occurs in AD, but AD brain global ubiquitin linkage profiling shows changes in other linkages as well [59]. Since K63 linked ubiquitin is not targeted to the proteasome, but does increase with V-ATPase acidification in the model of lysosomal insufficiency, the increase in K63 linkages seen in AD implicates accumulation of ubiquitinated proteins with obligate ESCRT-mediated degradation. Thus, trafficking, inflammatory signaling, and cell-type specific roles of dynamic lysosomal acidification are becoming increasingly appreciated for potential roles in AD pathogenesis.
Finally, it is important to point out that the general processes of endocytosis and endosomal-lysosomal dysregulations above-mentioned, have profoundly distinct implications for potential functions associated with other neurodegenerative diseases, such as PD, ALS, and Frontotemporal lobe degeneration (FTLD).
Conclusions and perspective
The endosomal-lysosomal system is a complex and highly dynamic process, where internalized transmembrane proteins, receptors, receptor ligands, and some soluble extracellular proteins are transported, sorted, and/or degraded. In recent years, particular attention has been paid to the endosomal-lysosomal system because it is involved in almost all of the neurodegenerative diseases, even though how it does so in each still remains unclear. Ongoing future studies will investigate both common and cell-type (or even local membrane region) specific trafficking and proteostasis pathways involving the endosomal-lysosomal system as well as the larger endomembrane system.
For example, a better understanding of distinct roles that ubiquitination plays in ESCRT-mediated proteostasis (and even lipid droplet homeostasis [79] which appears to be dysregulated in glia in neurodegeneration [47, 80]) could help to predict and ultimately therapeutically address the onset and progression of neurodegenerative diseases for specific individuals or sub-populations. This milieu of membrane-bound proteins that dynamically sorts cargo enriched for signaling, inflammation, and neurotrophic functions—among others—promises to provide a mother lode of new therapeutic targets for amelioriating neurodegenerative diseases, but the exploration also promises to be challenging, requiring the development of novel techniques and insight.
Within the secreted phospholipase A2 (sPLA2) family, group X sPLA2 (sPLA2-X) has the highest capacity to hydrolyze cellular membranes and has long been thought to promote inflammation by releasing arachidonic acid, a precursor of pro-inflammatory eicosanoids. Unexpectedly, we found that transgenic mice globally overexpressing human sPLA2-X (PLA2G10-Tg) displayed striking immunosuppressive and lean phenotypes with lymphopenia and increased M2-like macrophages, accompanied by marked elevation of free ω3 polyunsaturated fatty acids (PUFAs) and their metabolites. Studies using Pla2g10-deficient mice revealed that endogenous sPLA2-X, which is highly expressed in the colon epithelium and spermatozoa, mobilized ω3 PUFAs or their metabolites to protect against dextran sulfate-induced colitis and to promote fertilization, respectively. In colitis, sPLA2-X deficiency increased colorectal expression of Th17 cytokines, and ω3 PUFAs attenuated their production by lamina propria cells partly through the fatty acid receptor GPR120. In comparison, cytosolic phospholipase A2 (cPLA2α) protects from colitis by mobilizing ω6 arachidonic acid metabolites, including prostaglandin E2. Thus, our results underscore a previously unrecognized role of sPLA2-X as an ω3 PUFA mobilizer in vivo, segregated mobilization of ω3 and ω6 PUFA metabolites by sPLA2-X and cPLA2α, respectively, in protection against colitis, and the novel role of a particular sPLA2-X-driven PUFA in fertilization.
ChooseLife : Yet more evidence, of the beneficial properties imbued in individuals, who choose to pay head to Budwig et al. and seek to gain a better balance in their Omega3 to Omega6 ratios. I wouldn’t be without my Flaxoil/Flaxseeds, neither would my… too much!