
Published : 2015 Sep 30. doi: 10.1186/s40035-015-0041-1
Authors : Yong-Bo Hu Eric B Dammer Ru-Jing Ren Gang Wang
Abstract
The endosomal-lysosomal system is made up of a set of intracellular membranous compartments that dynamically interconvert, which is comprised of early endosomes, recycling endosomes, late endosomes, and the lysosome. In addition, autophagosomes execute autophagy, which delivers intracellular contents to the lysosome. Maturation of endosomes and/or autophagosomes into a lysosome creates an unique acidic environment within the cell for proteolysis and recycling of unneeded cellular components into usable amino acids and other biomolecular building blocks.
In the endocytic pathway, gradual maturation of endosomes into a lysosome and acidification of the late endosome are accompanied by vesicle trafficking, protein sorting and targeted degradation of some sorted cargo. Two opposing sorting systems are operating in these processes: the endosomal sorting complex required for transport (ESCRT) supports targeted degradation and the retromer supports retrograde retrieval of certain cargo.
The endosomal-lysosomal system is emerging as a central player in a host of neurodegenerative diseases, demonstrating potential roles which are likely to be revealed in pathogenesis and for viable therapeutic strategies. Here we focus on the physiological process of endosomal-lysosomal maturation, acidification and sorting systems along the endocytic pathway, and further discuss relationships between abnormalities in the endosomal-lysosomal system and neurodegenerative diseases, especially Alzheimer’s disease (AD).
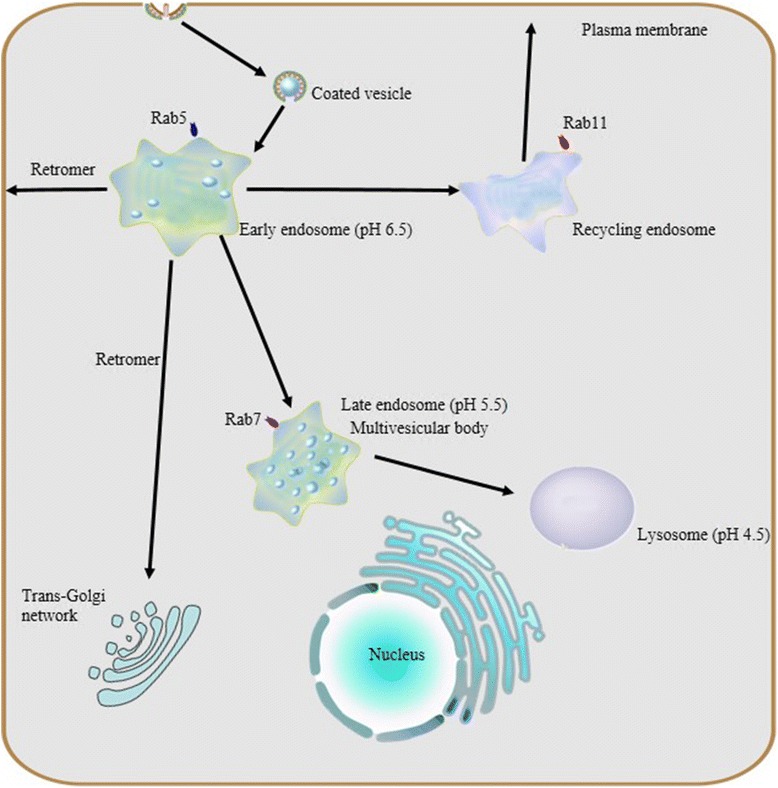
The endosomal-lysosomal system is a series of organelles in the endocytic pathway where various cargo molecules required for normal cellular function are internalized, recycled and modulated. Recently, mounting evidence has suggested that abnormalities in both endosomes and lysosomes, or dysregulation in their trafficking, play an important role directly in a surprising host of neurological dysfunctions, represented by AD, Parkinson’s disease (PD), and Lewy body dementia (LBD) [1–3]. Thus, the endosomal-lysosomal system is emerging as a key to understanding the mechanisms underlying both protein degradation and neurodegeneration. Here, we intend to summarize advances in the study of the endosomal-lysosomal system, with a focus on compartmentalized organization of trafficking routes, sorting machinery and their relationships to neurodegeneration.
A pH gradient established by vacuolar ATPase and chloride channels
Vacuolar ATP-dependent acidification
As mentioned above, the endosomal and lysosomal compartments share at least one similar significant characteristic: low intraluminal pH. These sealed acidic compartments provide an appropriate environment for optimal degradation of substrate cargo and recycling of their cognate receptors. As macromolecules are transported along the endosomal-lysosomal pathway, the internal pH of both endosomes and lysosomes decreases rapidly due to vacuolar acidification.
Previous research revealed that the same H+-ATPase, also known as vacuolar ATPase (V-ATPase), acidifies both endosomes and lysosomes. V-ATPase, differs from Na+,K+-ATPase in the plasma membrane, Ca2+-ATPase in the sarcoplasmic reticulum, and F1,F0-ATP synthase in mitochondria, in that it does not require a coupled influx of permeant anions [19]. Sulfhydryl alkylating reagents such as N-ethylmaleimide inhibit the V-ATPase dependent acidification of the endosomal-lysosomal system [19–21] as well as the specific inhibitor Bafilomycin A1 [22, 23].
V-ATPase is a unique class of ATPase present throughout the membranes which constrain the endocytic pathway, including the trans golgi network (TGN). V-ATPase, as a protein complex, is composed of two multimeric subunits, V1 in the cytoplasmic domain and V0 within the vacuolar membrane; the activity of V-ATPase depends on the dynamic assembly of these. V-ATPase is widely expressed in eukaryotic cells and serves as the master regulator of vesicular acidification in many subcellular membrane bound organelles. It also has important roles to play in vesicular trafficking and proteostasis.
Abnormalities and dysregulation of the endosomal-lysosomal system in neurodegeneration
Increasing attention being paid to the endosomal-lysosomal system has begun to elucidate a relationship between endosomal-lysosomal defects and neurodegeneration. In particular, robust pathology implicating endosomal-lysosomal disruption in AD has been well characterized. Here, we focus preferably on AD as a example of neurodegenerative disease and we believe that AD represents a general model of neurodegenerative diseases on abnormalities of the endosomal-lysosomal system occur along a continuum that includes early endosome changes, dysregulated acidification and sorting component defects.
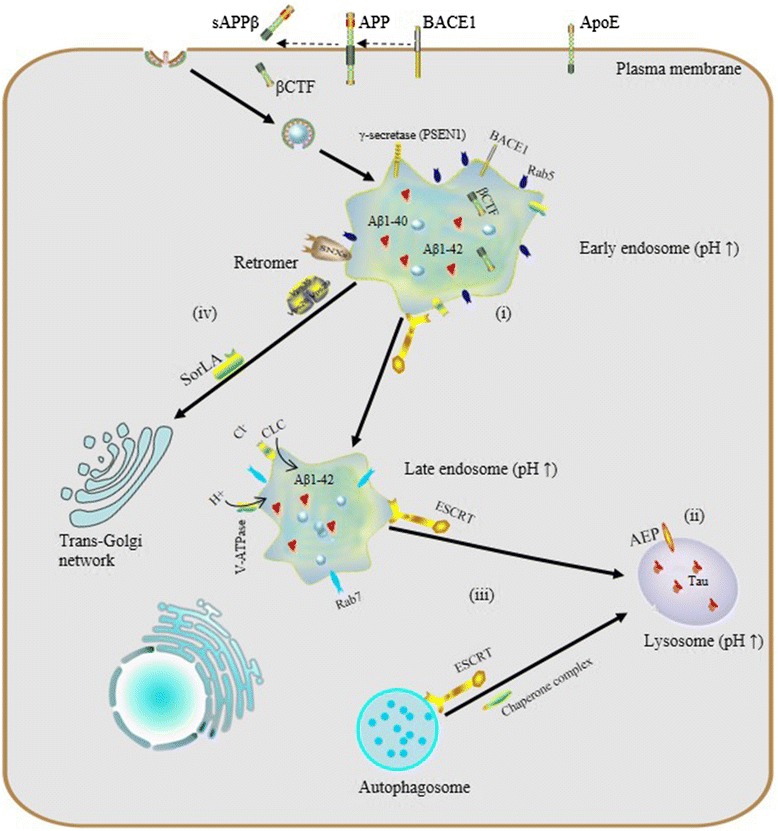
Dysregulated acidification, cellular indigestion?
Endosomal-lysosomal pH defects are an emerging theme in mechanisms underlying a number of neurodegenerative diseases. To date, results from experiments in vivo and in vitro have revealed the importance of proper vesicular pH balance and optimal acidification in transporting and degrading cargo via the endocytic pathway [48, 49]. For instance, Lee et al. reported that, in presenilin1 (PSEN1)-deleted blastocysts, defective lysosome acidification was observed with a substaintially elevated lysosomal PH of 5.4 and PSEN1 is essential for the transport of mature V0a1 subunites of V-ATP to lysosomes for their acidification and proteolysis [50].
Specifically, dysregulation of acidification and intracellular pH perturbation could influence the activity of enzymes in endomembrane compartments, resulting in impaired clearance of protein aggregates downstream of elevated endomembrane system pH, or conversely, due to decreased cytoplasmic pH. Regarding the latter, asparaginyl endopeptidase (AEP) is a typical pH-sensitive protein hydrolase the activity of which depends on the acidic pH of vesicular compartments. Predominantly localized in late endosomes, asparaginyl endopeptidase (AEP) specifically cleaves substrates with an asparagine residue at the P1 site. It is known that AEP can undergo reversible pH-dependent autoproteolytic activation, and in normal conditions, full-length pro-AEP is inactive [51].
As pH decreases from neutral to acidic, the activity of AEP gradually increases, such that it is partially activated at pH 4.5 and fully activated at pH 3.5, via removal of a cap that covers the active site. In AD patients, lysosomal acidification may be defective and it has been shown that the intracellular pH of neurons gradually decreases with aging [52] and more so with lactic acid elevation seen in AD cortex [53], so ectopic AEP activation or activity after leakage of active enzyme from late endosomes or lysosomes may be increased. AEP is involved in pathological tau degradation. Specifically, AEP generates tau fragments that form insoluble fibrils and result in neurotoxicity and neuropathological changes in AD [54, 55].
Increased endosome and lysosome pH is expected to have global effects on the proteome, particularly membrane proteins which rely on this pathway for their regulation and degradation. Interestingly, studies of microglia in culture have shown that in the absence of inflammatory IL-6 signaling, microglia do not achieve a sufficiently low lysosomal pH to degrade Aß, while after stimulation, CLC7 trafficking to lysosomes increases and pH drops sufficiently to improve Aß clearance [49, 56].
We have recently performed a systematic look at the proteomic effects of defective endosomal-lysosomal pH in a cellular model, in order to develop a better understanding of the global changes in the proteome that follow inhibition of V-ATPase and could be considered together as a signature or biomarker of defective vacuolar acidification [57, 58], which would be expected to have an overlap with changes seen in AD and/or other conditions which may be subject to this often age-dependent defect.
Indeed, blocking lysosomal degradation with bafilomycin A1 affects a significant increase in global K63 polyubiquitin linkages, which also occurs in AD, but AD brain global ubiquitin linkage profiling shows changes in other linkages as well [59]. Since K63 linked ubiquitin is not targeted to the proteasome, but does increase with V-ATPase acidification in the model of lysosomal insufficiency, the increase in K63 linkages seen in AD implicates accumulation of ubiquitinated proteins with obligate ESCRT-mediated degradation. Thus, trafficking, inflammatory signaling, and cell-type specific roles of dynamic lysosomal acidification are becoming increasingly appreciated for potential roles in AD pathogenesis.
Finally, it is important to point out that the general processes of endocytosis and endosomal-lysosomal dysregulations above-mentioned, have profoundly distinct implications for potential functions associated with other neurodegenerative diseases, such as PD, ALS, and Frontotemporal lobe degeneration (FTLD).
Conclusions and perspective
The endosomal-lysosomal system is a complex and highly dynamic process, where internalized transmembrane proteins, receptors, receptor ligands, and some soluble extracellular proteins are transported, sorted, and/or degraded. In recent years, particular attention has been paid to the endosomal-lysosomal system because it is involved in almost all of the neurodegenerative diseases, even though how it does so in each still remains unclear. Ongoing future studies will investigate both common and cell-type (or even local membrane region) specific trafficking and proteostasis pathways involving the endosomal-lysosomal system as well as the larger endomembrane system.
For example, a better understanding of distinct roles that ubiquitination plays in ESCRT-mediated proteostasis (and even lipid droplet homeostasis [79] which appears to be dysregulated in glia in neurodegeneration [47, 80]) could help to predict and ultimately therapeutically address the onset and progression of neurodegenerative diseases for specific individuals or sub-populations. This milieu of membrane-bound proteins that dynamically sorts cargo enriched for signaling, inflammation, and neurotrophic functions—among others—promises to provide a mother lode of new therapeutic targets for amelioriating neurodegenerative diseases, but the exploration also promises to be challenging, requiring the development of novel techniques and insight.
Full Paper : https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4596472/